Scoring
Mascot Probability Based Scoring
Mascot uses probability based scoring. This enables a simple rule to be used to judge whether a result is significant or not.
Matches using mass values (either peptide masses or MS/MS fragment ion masses) are always handled on a probabilistic basis. The total score is the probability that the observed match is a random event. Reporting probabilities directly can be confusing. Partly because they encompass a very wide range of magnitudes, and also because a "high" score is a "low" probability, which can be ambiguous. For this reason, we report scores as -10*LOG10(P), where P is the absolute probability. A probability of 10-20 thus becomes a score of 200.
Significance Level
A commonly accepted threshold is that an event is significant if it would be expected to occur at random with a frequency of less than 5%. This is the default value that is reported on the results summary page.
The Protein Summary page for typical peptide mass fingerprint search (open in new window) reports that "Scores greater than 70 are significant (p<0.05)". The histogram of the score distribution looks like this:
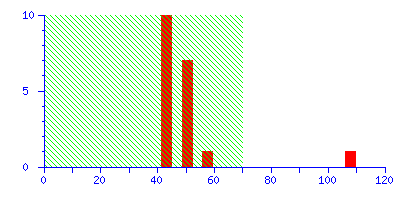
The protein with the high score of 108 is a 26 kDa heat shock protein from yeast. This is a nice result because the highest score is highly significant, leaving little room for doubt.
(It may be useful to think of the score histogram as a highly magnified view of the extreme tail of the distribution of scores for all the entries in the sequence database. In this case, 50 entries out of 561,356. Scores in the green region are inside this tail, and are of no significance. A real match, which is a non-random event, gives a score which is well clear of the tail.)
It is important to distinguish between a significant match and the best match. Ideally, the correct match is both the best match and a significant match. However, significance is a function of data quality. It may be that there are just not enough mass values or the mass measurement accuracy is not good enough to get a significant match. This doesn’t mean that the best match isn’t correct, it just means that you must study the result more critically.
To illustrate the difference between a significant match and a correct match, try repeating the search in the example, but with the mass tolerance increased from ±0.1 Da to ±1.0 Da. The discrimination of the search is greatly reduced, and the score for the correct match falls just below the significance level:
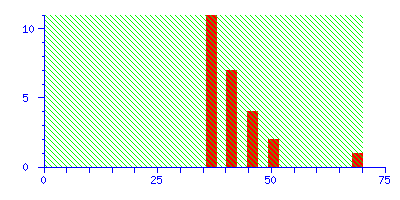
The best match is still correct, but it not significant. If we did 20 such searches, we could expect to get this score by chance alone because there is such a huge number of entries in the sequence database. Increase the mass tolerance to ±2.0 Da, and the correct match is no longer the protein with the highest score.
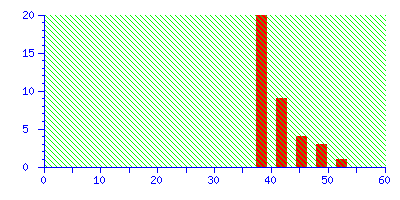
Even if this was an unknown, it is clear from the significance level that this is not a useful match, and there is no danger of this result becoming a false positive.
Expectation Values
Each protein score in a peptide mass fingerprint, and each ions score in an MS/MS search, is accompanied by an expectation value. This is the number of matches with equal or better scores that are expected to occur by chance alone. It is directly equivalent to the E-value in a Blast search result. For a score that is exactly on the default significance threshold, (p<0.05), the expectation value is also 0.05. Increase the score by 10 and the expectation value drops to 0.005. The lower the expectation value, the more significant the score.
Mass Tolerances
If the number of matched mass values is constant, the score in a peptide mass fingerprint will be inversely related to the mass tolerance, as shown in the example above. This is not the case for an MS/MS ions search, where increasing the peptide mass tolerance will have no effect on the ions score. This is because the ions score comes from the MS/MS fragment ion matches. Opening up the peptide mass tolerance means that Mascot has to test many more peptides, so the search takes longer and the discrimination is reduced, but the ions score remains unchanged.
Of course, if the peptide mass tolerance is set too tightly, in an effort to improve discrimination, one or more of the peptide matches may be lost, which will dramatically reduce the overall score.
Limitations
Like any statistical approach, Probability Based scoring depends on assumptions and models.
One of these assumptions is that the entries in the sequence databases can be modelled as random sequences. This is not always a good assumption. Some of the most glaring examples involve extended repeats, such as AAC62527, porcine submaxillary apomucin. Although the molecular weight of this protein is 1.2 MDa, over 80% of the sequence is composed of an identical 7 kDa repeat. It is difficult to know how to treat such cases. If a single experimental peptide mass is allowed to match to multiple calculated masses, then a single experimental mass which matches within a repeat will give a huge and meaningless score. But, if duplicate matches are not permitted, it will be virtually impossible to get a match to such a protein because the number of measurable mass values is too small to give a statistically significant score.
Another assumption is that the experimental measurements are independent determinations. This will not be true if the data include multiple mass values for the same peptide, even if these are from ions with different charge states in an electrospray LC-MS run. Good peak detection and thresholding (in both mass and time domains for LC-MS) are essential for any scoring algorithm to give meaningful results.
Sequence Query Scoring
Amino acid sequence or composition information, if included as seq(…) or comp(…) qualifiers, is treated as a filter on the candidate sequences. Ambiguous sequence or composition data can be used (in a manner similar to a regular expression search in computing) but it still functions as a filter, not a probabilistic match of the type found in a Blast or Fasta search.
In contrast, tag(…) and etag(…) qualifiers are scored probabilistically. That is, the more qualifiers that match, the higher the score, but all qualifiers are not required to match.
